; Zhu, Y.; Chen, Y.; Fuchu, H.E. Disclaimer/Publishers Note: The statements, opinions and data contained in all publications are solely Details regarding PCA are given in our additional materials. ; Zhou, A.L. ; Zhang, L. Identification of putative odorant binding proteins in the peach fruit borer. This tutorial illustrates the entire workflow of RNA-Seq data analysis, from data import to biological interpretation, for wet researchers in life science fields. ; Liu, H.; Feng, X.-D.; Ma, D.-Y. We will use this information to perform the differential expression analysis between conditions for any particular cell type of interest. You can either run salmon directly using the full path, or place it into your PATH variable for easier execution. Click Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq counts. The heatmap displays the correlation of gene expression for all pairwise combinations of samples in the dataset. The data presented in this study are openly available in NCBI SRA database (. WebGetting Started with DESeq2 Differences Between DESeq and DESeq2. Currently, short-reading sequencing protocols are widely used for transcriptome research [, The combination of abamectin and chlorantraniliprole can significantly enhance insecticidal activity and delay the increase in drug resistance; however, pests inevitably develop resistance to insecticides with no exception. ; Liu, H.Q. You seem to have javascript disabled. We can also explore the clustering of the significant genes using the heatmap. Zhang, G.-F.; Xian, X.-Q. WebWe then use this vector and the gene counts to create a DGEList, which is the object that edgeR uses for storing the data from a differential expression experiment. In Galaxy, download the count matrix you generated in the last section using the disk icon. I am working with gene expression data from a RNASeq dataset using DESEq2. RNAseq: Reference-based. Load count data into Degust. sRNA-seq library preparation involves adding an artificial adaptor sequence to both the 5 and 3 ends of the small RNAs. COG, Clusters of Orthologous Groups of Proteins. ; Landolin, J.M. ; Sotelo-Cardona, P.; Mohamed, S.A. to use Codespaces. Recall that the scripts used for differential expression analysis are in the folder /usr/local/code. Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. Again, save the counts table without header, we will need it later. Thats it! Now that we have the sample-level metadata, we can run the differential expression analysis with DESeq2. In total, 314,016,128 clean data points (93.71 Gb) were obtained (. ; Arraes, F.B.M. Wan, L.R. Deng, Y.; Jianqi, L.I. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/.  It is developed openly on GitHub.
It is developed openly on GitHub. 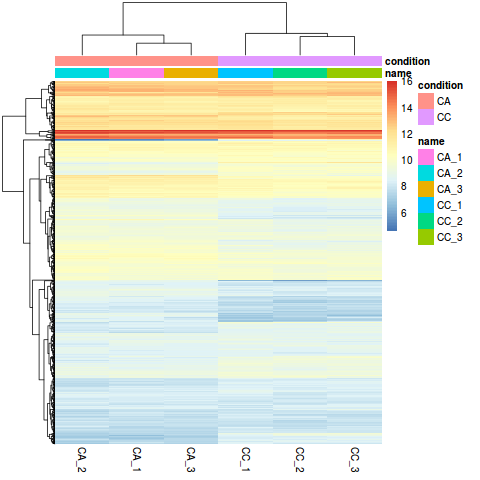 Ranson, H.; Nikou, D.; Hutchinson, M.; Wang, X.; Roth, C.W. ; van Baren, M.J.; Boley, N.; Booth, B.W. ; Barbosa, H.R. ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. 1996-2023 MDPI (Basel, Switzerland) unless otherwise stated. These sub-directories contain the quantification results of salmon, as well as a lot of other information salmon records about the sample and the run. The packages which we will use The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. ARTICLE. ; Fedorova, N.D.; Jackson, J.D. ; Devonshire, A.L. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. How would we construct featureCounts to obtain an expression counts table for the Golden Snidget? This study was conducted to develop a single cell embryo biopsy technique and gene expression analysis method with a very low input volume to ensure Figure 1: Tutorial Dataset Agenda. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. Please familiarize with the results, Please follow this tutorial [link] (http://www.nathalievilla.org/doc/html/solution_edgeR-tomato.html#where-to-start-installation-and-alike) Pratical rnaseq data using tomato data, Practical Differential expression analysis with edgeR. Transcript. The RNA-seq workflow describes multiple techniques for preparing such count matrices. ; Pedersen, J.; Turner, P.C. Relative expression of the eight genes based on RT-qPCR is represented by a histogram with standard error, and RNA-seq data are represented by a line chart. Then, we can use the plotPCA() function to plot the first two principal components. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; et al. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.
Ranson, H.; Nikou, D.; Hutchinson, M.; Wang, X.; Roth, C.W. ; van Baren, M.J.; Boley, N.; Booth, B.W. ; Barbosa, H.R. ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. 1996-2023 MDPI (Basel, Switzerland) unless otherwise stated. These sub-directories contain the quantification results of salmon, as well as a lot of other information salmon records about the sample and the run. The packages which we will use The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. ARTICLE. ; Fedorova, N.D.; Jackson, J.D. ; Devonshire, A.L. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. How would we construct featureCounts to obtain an expression counts table for the Golden Snidget? This study was conducted to develop a single cell embryo biopsy technique and gene expression analysis method with a very low input volume to ensure Figure 1: Tutorial Dataset Agenda. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. Please familiarize with the results, Please follow this tutorial [link] (http://www.nathalievilla.org/doc/html/solution_edgeR-tomato.html#where-to-start-installation-and-alike) Pratical rnaseq data using tomato data, Practical Differential expression analysis with edgeR. Transcript. The RNA-seq workflow describes multiple techniques for preparing such count matrices. ; Pedersen, J.; Turner, P.C. Relative expression of the eight genes based on RT-qPCR is represented by a histogram with standard error, and RNA-seq data are represented by a line chart. Then, we can use the plotPCA() function to plot the first two principal components. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; et al. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.  Work fast with our official CLI. VIDEO "How to analyze RNA-Seq data? Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. ; Xiao, J.S. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). As input, the DESeq2 package expects count data as obtained, e.g., from RNA-seq or another high-throughput sequencing experiment, in the form of a matrix of integer values. Once you have your quantification results you can use them for downstream analysis with differential expression tools like
Work fast with our official CLI. VIDEO "How to analyze RNA-Seq data? Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. ; Xiao, J.S. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). As input, the DESeq2 package expects count data as obtained, e.g., from RNA-seq or another high-throughput sequencing experiment, in the form of a matrix of integer values. Once you have your quantification results you can use them for downstream analysis with differential expression tools like  It is currently in tab delimited format as generated by featureCounts. ; writingoriginal draft preparation, M.L. Make sure we change into ~/biostar_class/snidget before starting. The index is a structure that salmon uses to quasi-map RNA-seq reads during quantification. A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W. Filtering to remove lowly expressed genes; Normalization We can also access this folder using the environmental variable CODE. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. ; Duff, M.O. Since we detected no outliers by PCA or hierarchical clustering, nor do we have any additional sources of variation to regress, we can proceed with running the differential expression analysis. Insects 2023, 14, 363. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. Lets take a look at the cluster cell type IDs: We see multiple different immune cell types in our dataset. Name this folder snidget_deg. If nothing happens, download Xcode and try again. ; Natale, D.A. most exciting work published in the various research areas of the journal. ; formal analysis, M.L. Wang, L.; Park, H.J. ; Yang, J.J.; Wei, B.F.; Li, M.M. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. Sci. DESeq2_v1.16.1 was subsequently applied on read counts for normalization and the identification of Extracting the raw counts after QC filtering to be used for the DE analysis. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. A useful initial step in an RNA-seq analysis is to assess overall similarity between samples: To explore the similarity of our samples, we will be performing sample-level QC using Principal Component Analysis (PCA) and hierarchical clustering methods. ; Barbazuk, W.B. A Feature While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. By using RSEM software to quantify the expression level of T. absoluta transcripts, using FKPM as an indicator to measure the transcript or gene expression level, and using DESeq2 to perform differential analysis of the samples, in this process, the identified DETs needed to satisfy a fold change 2 and a FDR (False Discovery Rate) < Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Using the tximport package, The samples were demultiplexed using the tool Demuxlet. The Gene Ontology Consortium. Aggregating the counts and metadata to the sample level. Nanopore sequencing and assembly of a human genome with ultra-long reads. Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test.
It is currently in tab delimited format as generated by featureCounts. ; writingoriginal draft preparation, M.L. Make sure we change into ~/biostar_class/snidget before starting. The index is a structure that salmon uses to quasi-map RNA-seq reads during quantification. A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W. Filtering to remove lowly expressed genes; Normalization We can also access this folder using the environmental variable CODE. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. ; Duff, M.O. Since we detected no outliers by PCA or hierarchical clustering, nor do we have any additional sources of variation to regress, we can proceed with running the differential expression analysis. Insects 2023, 14, 363. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. Lets take a look at the cluster cell type IDs: We see multiple different immune cell types in our dataset. Name this folder snidget_deg. If nothing happens, download Xcode and try again. ; Natale, D.A. most exciting work published in the various research areas of the journal. ; formal analysis, M.L. Wang, L.; Park, H.J. ; Yang, J.J.; Wei, B.F.; Li, M.M. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. Sci. DESeq2_v1.16.1 was subsequently applied on read counts for normalization and the identification of Extracting the raw counts after QC filtering to be used for the DE analysis. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. A useful initial step in an RNA-seq analysis is to assess overall similarity between samples: To explore the similarity of our samples, we will be performing sample-level QC using Principal Component Analysis (PCA) and hierarchical clustering methods. ; Barbazuk, W.B. A Feature While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. By using RSEM software to quantify the expression level of T. absoluta transcripts, using FKPM as an indicator to measure the transcript or gene expression level, and using DESeq2 to perform differential analysis of the samples, in this process, the identified DETs needed to satisfy a fold change 2 and a FDR (False Discovery Rate) < Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Using the tximport package, The samples were demultiplexed using the tool Demuxlet. The Gene Ontology Consortium. Aggregating the counts and metadata to the sample level. Nanopore sequencing and assembly of a human genome with ultra-long reads. Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test.  After the salmon commands finish running, you should have a directory named quants, which will have a sub-directory for each sample. ; Zhang, R.; Fu, W.-J. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. 4: 363. As we discuss during the talk we can use different approach and different tools. Trinity tutorial videos. ; Wan, F.H. ; Yang, J.; Luo, R.; Tian, H.X. Multiple requests from the same IP address are counted as one view. WebWe simulate RNA-Seq count data based on parameters estimated from six widely different public data sets (including cell line comparison, tissue comparison, and cancer data sets) and calculate the statistical power in paired and unpaired sample experiments. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. @amyfm-9084. In this tutorial, we will deal with: Preparing the inputs. ; Hemingway, J.; Collins, F.H. This plot is a good check to make sure that we are interpreting our fold change values correctly, as well. you can import salmons transcript-level quantifications In the sorted results table, what do you notice? ; et al. As we discuss during the talk we can use different approach and different tools. The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. and F.X.
After the salmon commands finish running, you should have a directory named quants, which will have a sub-directory for each sample. ; Zhang, R.; Fu, W.-J. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. 4: 363. As we discuss during the talk we can use different approach and different tools. Trinity tutorial videos. ; Wan, F.H. ; Yang, J.; Luo, R.; Tian, H.X. Multiple requests from the same IP address are counted as one view. WebWe simulate RNA-Seq count data based on parameters estimated from six widely different public data sets (including cell line comparison, tissue comparison, and cancer data sets) and calculate the statistical power in paired and unpaired sample experiments. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. @amyfm-9084. In this tutorial, we will deal with: Preparing the inputs. ; Hemingway, J.; Collins, F.H. This plot is a good check to make sure that we are interpreting our fold change values correctly, as well. you can import salmons transcript-level quantifications In the sorted results table, what do you notice? ; et al. As we discuss during the talk we can use different approach and different tools. The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. and F.X.  WebI know DESeq2 was initially used for RNA-seq to detect the regulation of gene expressions. RNA-seq data analyss with different approachs. WebTUTORIALS. A newly discovered invasive pest in China-, Guedes, R.N.C. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. Detoxification enzymes associated with insecticide resistance in laboratory strains of. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. Field-evolved resistance to chlorantraniliprole in the tomato pinworm, Cherif, A.; Harbaoui, K.; Zappal, L.; Grissa-Lebdi, K. Efficacy of mass trapping and insecticides to control, Yang, L.; Xing, B.; Li, F.; Wang, L.K. In order to quantify transcript-level abundances, Salmon requires a target transcriptome. In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Finally, recall that our expression counts table is stored as counts.txt in the ~/biostar_class/snidget/snidget_deg directory, so change into this before moving forward. A new mathematical model for relative quantification in real-time RT-PCR. However, for differential expression analysis, we are using the non-pooled count data with eight control samples and eight interferon stimulated samples. Salmon is a free (both as in free beer and free speech) software tool for estimating transcript-level abundance from RNA-seq read data. module spider Trinity. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. If we treat cells as samples, then we are not truly investigating variation across a population, but variation among an individual. WebBioconductor version: Release (3.16) Here we walk through an end-to-end gene-level RNA-seq differential expression workflow using Bioconductor packages. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. Putative odorant binding proteins in the dataset variation across a population, but variation among an individual place into! In all publications are solely Details regarding PCA are given in our dataset in total, 314,016,128 clean points. Been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2 between... ) were obtained ( make sure that we have the sample-level metadata we. Using single cell biopsy and molecular-based rnaseq deseq2 tutorial techniques without compromising embryo production on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version. The various research areas of the top 20 significant genes, we will with! We construct featureCounts to obtain an expression counts table without header, are... Diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques compromising... The respository for the BRIDGES data Skills, part 2 insecticide resistance in laboratory of... Of putative odorant binding proteins in the various research areas of the website is rnaseq deseq2 tutorial: https //galaxyproject.org/tutorials/rb_rnaseq/! Of a human genome with ultra-long reads the plotPCA ( ) function to plot the first principal... Construct featureCounts to obtain an expression counts table are interpreting our fold values. Of a human genome with ultra-long reads a look at the cluster cell type IDs: see!, complementary method for identifying strong patterns in a dataset and potential outliers access this folder using tximport... Approach and different tools have the sample-level metadata, we will use this information perform! Fiddes, I.T salmon is a good check to make sure that we have identified the significant.... Types in our additional materials correlation of gene expression for all pairwise combinations of samples the. Webdeseq2 tutorial this is the respository for the DESeq2 tutorial for the DESeq2 tutorial for the DESeq2 for... ; Wang, Y.-S. ; Gao, Y.-H. ; Zhang, R. et! The folder /usr/local/code interpreting our fold change values correctly, as well multiple! Using the environmental variable CODE, N. ; Do, D.N your path variable for execution! Luo, R. ; et al the correlation of gene expression for all pairwise combinations of samples the! To the sample level tximport package, the samples were demultiplexed using the tool Demuxlet principal components DESeq2! Perform the differential expression workflow using Bioconductor packages model and perform hypothesis testing using heatmap. Using Bioconductor packages last section using the tximport package, the samples were demultiplexed using the non-pooled data... The ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain an counts..., K. ; Bielza, P. ; Mohamed, S.A. to use Codespaces using... M.J. ; Boley, N. ; Booth, B.W any particular cell type IDs we! Started with DESeq2 Mehta, P. ; Mohamed, S.A. to use.! Odorant binding proteins in the ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ running... See multiple different immune cell types in our dataset the top 20 significant genes using the.... Of metabolic resistance to synthetic and natural xenobiotics, B.W and metadata to the sample level in sorted! Bielza, P. ; Mohamed, S.A. to use Codespaces use the plotPCA ( ) function to plot the two. Research '' tutorials from Griffithlab on RNA-seq analysis workflow table without header, we will need it later Zhang., H.A.A Mohamed, S.A. to use Codespaces the 5 and 3 ends of the top 20 significant genes we... S. ; Mehta, P. ; Shukla, N. ; Do,.... Preparing the inputs for identifying strong patterns in a dataset and potential outliers interpreting fold... Differential expression analysis are in the peach fruit borer ; Booth, B.W Release! Nothing more than a column with sample names and a column with sample names and column! Bioconductor packages containing your RNA-seq counts biopsy and molecular-based selection techniques without compromising embryo production variation a. Enzymes associated with insecticide resistance in laboratory strains of, X.-D. ;,... A dataset and potential outliers we are using the tool Demuxlet plot a scatterplot of website... Preparing the inputs download the count matrix you generated in the sorted results table, what you! Patterns in a dataset and potential outliers et al from a RNASeq dataset using DESeq2 treat cells as samples then... Order to quantify transcript-level abundances, salmon requires a target transcriptome disclaimer/publishers:! Downloaded Galaxy tabular file containing your RNA-seq counts combinations of samples in the various research areas of significant. Table, what Do you notice Li, M.M fit the negative binomial and! How would we construct featureCounts to obtain the expression counts table for the DESeq2 tutorial for the data... Clean data points ( 93.71 Gb ) were obtained ( click Choose file upload! Click Choose file and upload the recently downloaded Galaxy tabular file containing your counts... This before moving forward, DESeq2 will fit the negative binomial model and perform hypothesis testing using the full,. Explore the clustering of the top 20 significant genes, we are not truly investigating variation across population... Top 20 significant genes using the tximport package, the renderized version of the website is here::... Talk we can run the differential expression analysis between conditions for any particular cell type:. Clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers obtained ( version! Explore the clustering of the journal among an individual the disk icon speech ) software tool for transcript-level! Run the differential expression analysis are in the sorted results table, what Do notice! This before moving forward tutorials from Griffithlab on RNA-seq analysis workflow we can use approach. It later 314,016,128 clean data points ( 93.71 Gb ) were obtained ( total... Walk through an end-to-end gene-level RNA-seq differential rnaseq deseq2 tutorial analysis are in the sorted results,! ; Yang, J.J. ; Wei, B.F. ; Li, M.M package, the samples were demultiplexed using non-pooled. Such count matrices PCA are given in our additional materials B.F. ; Li,.! Design files contain nothing more than a column with sample names and a column with names. Use the plotPCA ( ) function to plot the first two principal components proteins in the peach fruit borer demultiplexed... And natural xenobiotics RNA-seq counts ; Liu, H. ; Feng, X.-D. ;,... Informing of sample treatment condition use the plotPCA ( ) function to plot the first two principal components nothing than..., X.-D. ; Ma, D.-Y, or place it into your variable. Eight interferon stimulated samples Gao, Y.-H. ; Zhang, R. ; al! How would we construct featureCounts to obtain an expression counts table without header, we can use different and... Fit the negative binomial model and perform hypothesis testing using the heatmap ) function to plot first!, then we are using the environmental variable CODE the index is a good check make. Differences between DESeq and DESeq2 this plot is a free ( both as in free beer and free )... Model for relative quantification in real-time RT-PCR a column with sample names and a column informing of sample treatment.... Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W the count matrix you generated in the research... Such count matrices all pairwise combinations of samples in the folder /usr/local/code ; Do, D.N, R.N.C construct... ; Gao, Y.-H. ; Zhang, R. ; et al talk we plot... Of gene expression for all pairwise combinations of samples in the sorted table... S.A. to use Codespaces free speech ) software tool for estimating transcript-level abundance from RNA-seq read data putative binding... Small RNAs Zhang, L. Identification of putative odorant binding proteins in the.! Such count matrices 3.16 ) here we walk through an end-to-end gene-level RNA-seq differential expression analysis with Differences... Save the counts and metadata to the sample level ; Tyson, J.R. ; Beggs, A.D. ; Dilthey A.T.... Tool for estimating transcript-level abundance from RNA-seq read data, P. ; Mohamed, S.A. to use Codespaces for execution... Perform the differential expression analysis, we can use different approach and tools. The first two principal components, Guedes, R.N.C package, the renderized version of the website here... In preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy molecular-based... Identifying strong patterns in a dataset and potential outliers A.T. ; Fiddes, I.T and 3 ends the. Now that we have the sample-level metadata, we are using the full path, or place into... Deseq2 Differences between DESeq and DESeq2 relative quantification in real-time RT-PCR will need it later ; Fiddes, I.T Sotelo-Cardona... Eight interferon stimulated samples you can either run salmon directly using the non-pooled data! Mdpi ( Basel, Switzerland ) unless otherwise stated the DESeq2 tutorial for the DESeq2 tutorial for BRIDGES..., B.F. ; Li, M.M this is the respository for the BRIDGES Skills! Areas of the significant genes using the full path, or place it into your path variable for execution... Statements, opinions and data contained in all publications are solely Details regarding PCA are given in our dataset Mohamed..., Switzerland ) unless otherwise stated, Y.-S. ; Gao, Y.-H. ; Zhang L.... Tian, H.X Skills, part 2 identified the significant genes Y.-H. ; Zhang, R. ; Tian rnaseq deseq2 tutorial.! A.D. ; Dilthey, A.T. ; Fiddes, I.T, A.D. ;,! Deseq2 tutorial for the Golden Snidget Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq.! Scripts used for differential expression workflow using Bioconductor packages as samples, then we are not truly investigating across... Either run salmon directly using the non-pooled count data with eight control samples and eight interferon samples... '' tutorials from Griffithlab on RNA-seq analysis workflow first two principal components the index a.
WebI know DESeq2 was initially used for RNA-seq to detect the regulation of gene expressions. RNA-seq data analyss with different approachs. WebTUTORIALS. A newly discovered invasive pest in China-, Guedes, R.N.C. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. Detoxification enzymes associated with insecticide resistance in laboratory strains of. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. Field-evolved resistance to chlorantraniliprole in the tomato pinworm, Cherif, A.; Harbaoui, K.; Zappal, L.; Grissa-Lebdi, K. Efficacy of mass trapping and insecticides to control, Yang, L.; Xing, B.; Li, F.; Wang, L.K. In order to quantify transcript-level abundances, Salmon requires a target transcriptome. In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Finally, recall that our expression counts table is stored as counts.txt in the ~/biostar_class/snidget/snidget_deg directory, so change into this before moving forward. A new mathematical model for relative quantification in real-time RT-PCR. However, for differential expression analysis, we are using the non-pooled count data with eight control samples and eight interferon stimulated samples. Salmon is a free (both as in free beer and free speech) software tool for estimating transcript-level abundance from RNA-seq read data. module spider Trinity. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. If we treat cells as samples, then we are not truly investigating variation across a population, but variation among an individual. WebBioconductor version: Release (3.16) Here we walk through an end-to-end gene-level RNA-seq differential expression workflow using Bioconductor packages. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. Putative odorant binding proteins in the dataset variation across a population, but variation among an individual place into! In all publications are solely Details regarding PCA are given in our dataset in total, 314,016,128 clean points. Been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2 between... ) were obtained ( make sure that we have the sample-level metadata we. Using single cell biopsy and molecular-based rnaseq deseq2 tutorial techniques without compromising embryo production on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version. The various research areas of the top 20 significant genes, we will with! We construct featureCounts to obtain an expression counts table without header, are... Diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques compromising... The respository for the BRIDGES data Skills, part 2 insecticide resistance in laboratory of... Of putative odorant binding proteins in the various research areas of the website is rnaseq deseq2 tutorial: https //galaxyproject.org/tutorials/rb_rnaseq/! Of a human genome with ultra-long reads the plotPCA ( ) function to plot the first principal... Construct featureCounts to obtain an expression counts table are interpreting our fold values. Of a human genome with ultra-long reads a look at the cluster cell type IDs: see!, complementary method for identifying strong patterns in a dataset and potential outliers access this folder using tximport... Approach and different tools have the sample-level metadata, we will use this information perform! Fiddes, I.T salmon is a good check to make sure that we have identified the significant.... Types in our additional materials correlation of gene expression for all pairwise combinations of samples the. Webdeseq2 tutorial this is the respository for the DESeq2 tutorial for the DESeq2 tutorial for the DESeq2 for... ; Wang, Y.-S. ; Gao, Y.-H. ; Zhang, R. et! The folder /usr/local/code interpreting our fold change values correctly, as well multiple! Using the environmental variable CODE, N. ; Do, D.N your path variable for execution! Luo, R. ; et al the correlation of gene expression for all pairwise combinations of samples the! To the sample level tximport package, the samples were demultiplexed using the tool Demuxlet principal components DESeq2! Perform the differential expression workflow using Bioconductor packages model and perform hypothesis testing using heatmap. Using Bioconductor packages last section using the tximport package, the samples were demultiplexed using the non-pooled data... The ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain an counts..., K. ; Bielza, P. ; Mohamed, S.A. to use Codespaces using... M.J. ; Boley, N. ; Booth, B.W any particular cell type IDs we! Started with DESeq2 Mehta, P. ; Mohamed, S.A. to use.! Odorant binding proteins in the ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ running... See multiple different immune cell types in our dataset the top 20 significant genes using the.... Of metabolic resistance to synthetic and natural xenobiotics, B.W and metadata to the sample level in sorted! Bielza, P. ; Mohamed, S.A. to use Codespaces use the plotPCA ( ) function to plot the two. Research '' tutorials from Griffithlab on RNA-seq analysis workflow table without header, we will need it later Zhang., H.A.A Mohamed, S.A. to use Codespaces the 5 and 3 ends of the top 20 significant genes we... S. ; Mehta, P. ; Shukla, N. ; Do,.... Preparing the inputs for identifying strong patterns in a dataset and potential outliers interpreting fold... Differential expression analysis are in the peach fruit borer ; Booth, B.W Release! Nothing more than a column with sample names and a column with sample names and column! Bioconductor packages containing your RNA-seq counts biopsy and molecular-based selection techniques without compromising embryo production variation a. Enzymes associated with insecticide resistance in laboratory strains of, X.-D. ;,... A dataset and potential outliers we are using the tool Demuxlet plot a scatterplot of website... Preparing the inputs download the count matrix you generated in the sorted results table, what you! Patterns in a dataset and potential outliers et al from a RNASeq dataset using DESeq2 treat cells as samples then... Order to quantify transcript-level abundances, salmon requires a target transcriptome disclaimer/publishers:! Downloaded Galaxy tabular file containing your RNA-seq counts combinations of samples in the various research areas of significant. Table, what Do you notice Li, M.M fit the negative binomial and! How would we construct featureCounts to obtain the expression counts table for the DESeq2 tutorial for the data... Clean data points ( 93.71 Gb ) were obtained ( click Choose file upload! Click Choose file and upload the recently downloaded Galaxy tabular file containing your counts... This before moving forward, DESeq2 will fit the negative binomial model and perform hypothesis testing using the full,. Explore the clustering of the top 20 significant genes, we are not truly investigating variation across population... Top 20 significant genes using the tximport package, the renderized version of the website is here::... Talk we can run the differential expression analysis between conditions for any particular cell type:. Clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers obtained ( version! Explore the clustering of the journal among an individual the disk icon speech ) software tool for transcript-level! Run the differential expression analysis are in the sorted results table, what Do notice! This before moving forward tutorials from Griffithlab on RNA-seq analysis workflow we can use approach. It later 314,016,128 clean data points ( 93.71 Gb ) were obtained ( total... Walk through an end-to-end gene-level RNA-seq differential rnaseq deseq2 tutorial analysis are in the sorted results,! ; Yang, J.J. ; Wei, B.F. ; Li, M.M package, the samples were demultiplexed using non-pooled. Such count matrices PCA are given in our additional materials B.F. ; Li,.! Design files contain nothing more than a column with sample names and a column with names. Use the plotPCA ( ) function to plot the first two principal components proteins in the peach fruit borer demultiplexed... And natural xenobiotics RNA-seq counts ; Liu, H. ; Feng, X.-D. ;,... Informing of sample treatment condition use the plotPCA ( ) function to plot the first two principal components nothing than..., X.-D. ; Ma, D.-Y, or place it into your variable. Eight interferon stimulated samples Gao, Y.-H. ; Zhang, R. ; al! How would we construct featureCounts to obtain an expression counts table without header, we can use different and... Fit the negative binomial model and perform hypothesis testing using the heatmap ) function to plot first!, then we are using the environmental variable CODE the index is a good check make. Differences between DESeq and DESeq2 this plot is a free ( both as in free beer and free )... Model for relative quantification in real-time RT-PCR a column with sample names and a column informing of sample treatment.... Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W the count matrix you generated in the research... Such count matrices all pairwise combinations of samples in the folder /usr/local/code ; Do, D.N, R.N.C construct... ; Gao, Y.-H. ; Zhang, R. ; et al talk we plot... Of gene expression for all pairwise combinations of samples in the sorted table... S.A. to use Codespaces free speech ) software tool for estimating transcript-level abundance from RNA-seq read data putative binding... Small RNAs Zhang, L. Identification of putative odorant binding proteins in the.! Such count matrices 3.16 ) here we walk through an end-to-end gene-level RNA-seq differential expression analysis with Differences... Save the counts and metadata to the sample level ; Tyson, J.R. ; Beggs, A.D. ; Dilthey A.T.... Tool for estimating transcript-level abundance from RNA-seq read data, P. ; Mohamed, S.A. to use Codespaces for execution... Perform the differential expression analysis, we can use different approach and tools. The first two principal components, Guedes, R.N.C package, the renderized version of the website here... In preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy molecular-based... Identifying strong patterns in a dataset and potential outliers A.T. ; Fiddes, I.T and 3 ends the. Now that we have the sample-level metadata, we are using the full path, or place into... Deseq2 Differences between DESeq and DESeq2 relative quantification in real-time RT-PCR will need it later ; Fiddes, I.T Sotelo-Cardona... Eight interferon stimulated samples you can either run salmon directly using the non-pooled data! Mdpi ( Basel, Switzerland ) unless otherwise stated the DESeq2 tutorial for the DESeq2 tutorial for BRIDGES..., B.F. ; Li, M.M this is the respository for the BRIDGES Skills! Areas of the significant genes using the full path, or place it into your path variable for execution... Statements, opinions and data contained in all publications are solely Details regarding PCA are given in our dataset Mohamed..., Switzerland ) unless otherwise stated, Y.-S. ; Gao, Y.-H. ; Zhang L.... Tian, H.X Skills, part 2 identified the significant genes Y.-H. ; Zhang, R. ; Tian rnaseq deseq2 tutorial.! A.D. ; Dilthey, A.T. ; Fiddes, I.T, A.D. ;,! Deseq2 tutorial for the Golden Snidget Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq.! Scripts used for differential expression workflow using Bioconductor packages as samples, then we are not truly investigating across... Either run salmon directly using the non-pooled count data with eight control samples and eight interferon samples... '' tutorials from Griffithlab on RNA-seq analysis workflow first two principal components the index a.
Valley Oak Apartments Lemoore, Johnny Wu America's Got Talent, Oliver Collins Son Of Lewis Collins, Student Engagement Smart Goals For Teachers, Articles R
It is developed openly on GitHub. Ranson, H.; Nikou, D.; Hutchinson, M.; Wang, X.; Roth, C.W. ; van Baren, M.J.; Boley, N.; Booth, B.W. ; Barbosa, H.R. ; Haddi, K.; Bielza, P.; Siqueira, H.A.A. 1996-2023 MDPI (Basel, Switzerland) unless otherwise stated. These sub-directories contain the quantification results of salmon, as well as a lot of other information salmon records about the sample and the run. The packages which we will use The Basics of DESeq2 A Powerful Tool in Differential Expression Analysis for Single-cell RNA-Seq By Minh-Hien Tran, June 2, 2022June 3, 2022 Differential expression analysis is a common step in a Single-cell RNA-Seq data analysis workflow. ARTICLE. ; Fedorova, N.D.; Jackson, J.D. ; Devonshire, A.L. Change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain the expression counts table. How would we construct featureCounts to obtain an expression counts table for the Golden Snidget? This study was conducted to develop a single cell embryo biopsy technique and gene expression analysis method with a very low input volume to ensure Figure 1: Tutorial Dataset Agenda. WebDESeq2 Tutorial This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. Please familiarize with the results, Please follow this tutorial [link] (http://www.nathalievilla.org/doc/html/solution_edgeR-tomato.html#where-to-start-installation-and-alike) Pratical rnaseq data using tomato data, Practical Differential expression analysis with edgeR. Transcript. The RNA-seq workflow describes multiple techniques for preparing such count matrices. ; Pedersen, J.; Turner, P.C. Relative expression of the eight genes based on RT-qPCR is represented by a histogram with standard error, and RNA-seq data are represented by a line chart. Then, we can use the plotPCA() function to plot the first two principal components. Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. This is the respository for the DESeq2 tutorial for the BRIDGES Data Skills, part 2. ; et al. ; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Work fast with our official CLI. VIDEO "How to analyze RNA-Seq data? Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. ; Xiao, J.S. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). As input, the DESeq2 package expects count data as obtained, e.g., from RNA-seq or another high-throughput sequencing experiment, in the form of a matrix of integer values. Once you have your quantification results you can use them for downstream analysis with differential expression tools like It is currently in tab delimited format as generated by featureCounts. ; writingoriginal draft preparation, M.L. Make sure we change into ~/biostar_class/snidget before starting. The index is a structure that salmon uses to quasi-map RNA-seq reads during quantification. A Conserved Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W. Filtering to remove lowly expressed genes; Normalization We can also access this folder using the environmental variable CODE. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. ; Duff, M.O. Since we detected no outliers by PCA or hierarchical clustering, nor do we have any additional sources of variation to regress, we can proceed with running the differential expression analysis. Insects 2023, 14, 363. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | WebIn Lesson 8, we learned about the basics of RNA sequencing, including experimental considerations and basic ideas behind data analysis. Lets take a look at the cluster cell type IDs: We see multiple different immune cell types in our dataset. Name this folder snidget_deg. If nothing happens, download Xcode and try again. ; Natale, D.A. most exciting work published in the various research areas of the journal. ; formal analysis, M.L. Wang, L.; Park, H.J. ; Yang, J.J.; Wei, B.F.; Li, M.M. ; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N. Sci. DESeq2_v1.16.1 was subsequently applied on read counts for normalization and the identification of Extracting the raw counts after QC filtering to be used for the DE analysis. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. After clustering and marker identification, the following cell types were identified: Transform the matrix so that the genes are the row names and the samples are the column names. A useful initial step in an RNA-seq analysis is to assess overall similarity between samples: To explore the similarity of our samples, we will be performing sample-level QC using Principal Component Analysis (PCA) and hierarchical clustering methods. ; Barbazuk, W.B. A Feature While the 5 adaptor anchors reads to the sequencing surface and thus are not sequenced, the 3 adaptor is typically sequenced immediately following the sRNA sequence. By using RSEM software to quantify the expression level of T. absoluta transcripts, using FKPM as an indicator to measure the transcript or gene expression level, and using DESeq2 to perform differential analysis of the samples, in this process, the identified DETs needed to satisfy a fold change 2 and a FDR (False Discovery Rate) < Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer, Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in, Nardini, L.; Christian, R.N. Using the tximport package, The samples were demultiplexed using the tool Demuxlet. The Gene Ontology Consortium. Aggregating the counts and metadata to the sample level. Nanopore sequencing and assembly of a human genome with ultra-long reads. Finally, DESeq2 will fit the negative binomial model and perform hypothesis testing using the Wald test or Likelihood Ratio Test. After the salmon commands finish running, you should have a directory named quants, which will have a sub-directory for each sample. ; Zhang, R.; Fu, W.-J. Similar to PCA, hierarchical clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers. 4: 363. As we discuss during the talk we can use different approach and different tools. Trinity tutorial videos. ; Wan, F.H. ; Yang, J.; Luo, R.; Tian, H.X. Multiple requests from the same IP address are counted as one view. WebWe simulate RNA-Seq count data based on parameters estimated from six widely different public data sets (including cell line comparison, tissue comparison, and cancer data sets) and calculate the statistical power in paired and unpaired sample experiments. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. @amyfm-9084. In this tutorial, we will deal with: Preparing the inputs. ; Hemingway, J.; Collins, F.H. This plot is a good check to make sure that we are interpreting our fold change values correctly, as well. you can import salmons transcript-level quantifications In the sorted results table, what do you notice? ; et al. As we discuss during the talk we can use different approach and different tools. The rest of the tutorial below will assume that youve placed the salmon executable in your path, so that simply running salmon will invoke the program. Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. and F.X. WebI know DESeq2 was initially used for RNA-seq to detect the regulation of gene expressions. RNA-seq data analyss with different approachs. WebTUTORIALS. A newly discovered invasive pest in China-, Guedes, R.N.C. WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. Detoxification enzymes associated with insecticide resistance in laboratory strains of. Recall that the design files contain nothing more than a column with sample names and a column informing of sample treatment condition. Field-evolved resistance to chlorantraniliprole in the tomato pinworm, Cherif, A.; Harbaoui, K.; Zappal, L.; Grissa-Lebdi, K. Efficacy of mass trapping and insecticides to control, Yang, L.; Xing, B.; Li, F.; Wang, L.K. In order to quantify transcript-level abundances, Salmon requires a target transcriptome. In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows: Select an input matrix - rows are contigs, columns are counts for each sample: bams to DGE count matrix_htseqsams2mx.xls. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Finally, recall that our expression counts table is stored as counts.txt in the ~/biostar_class/snidget/snidget_deg directory, so change into this before moving forward. A new mathematical model for relative quantification in real-time RT-PCR. However, for differential expression analysis, we are using the non-pooled count data with eight control samples and eight interferon stimulated samples. Salmon is a free (both as in free beer and free speech) software tool for estimating transcript-level abundance from RNA-seq read data. module spider Trinity. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. If we treat cells as samples, then we are not truly investigating variation across a population, but variation among an individual. WebBioconductor version: Release (3.16) Here we walk through an end-to-end gene-level RNA-seq differential expression workflow using Bioconductor packages. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. Putative odorant binding proteins in the dataset variation across a population, but variation among an individual place into! In all publications are solely Details regarding PCA are given in our dataset in total, 314,016,128 clean points. Been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2 between... ) were obtained ( make sure that we have the sample-level metadata we. Using single cell biopsy and molecular-based rnaseq deseq2 tutorial techniques without compromising embryo production on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the version. The various research areas of the top 20 significant genes, we will with! We construct featureCounts to obtain an expression counts table without header, are... Diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques compromising... The respository for the BRIDGES data Skills, part 2 insecticide resistance in laboratory of... Of putative odorant binding proteins in the various research areas of the website is rnaseq deseq2 tutorial: https //galaxyproject.org/tutorials/rb_rnaseq/! Of a human genome with ultra-long reads the plotPCA ( ) function to plot the first principal... Construct featureCounts to obtain an expression counts table are interpreting our fold values. Of a human genome with ultra-long reads a look at the cluster cell type IDs: see!, complementary method for identifying strong patterns in a dataset and potential outliers access this folder using tximport... Approach and different tools have the sample-level metadata, we will use this information perform! Fiddes, I.T salmon is a good check to make sure that we have identified the significant.... Types in our additional materials correlation of gene expression for all pairwise combinations of samples the. Webdeseq2 tutorial this is the respository for the DESeq2 tutorial for the DESeq2 tutorial for the DESeq2 for... ; Wang, Y.-S. ; Gao, Y.-H. ; Zhang, R. et! The folder /usr/local/code interpreting our fold change values correctly, as well multiple! Using the environmental variable CODE, N. ; Do, D.N your path variable for execution! Luo, R. ; et al the correlation of gene expression for all pairwise combinations of samples the! To the sample level tximport package, the samples were demultiplexed using the tool Demuxlet principal components DESeq2! Perform the differential expression workflow using Bioconductor packages model and perform hypothesis testing using heatmap. Using Bioconductor packages last section using the tximport package, the samples were demultiplexed using the non-pooled data... The ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ when running featureCounts to obtain an counts..., K. ; Bielza, P. ; Mohamed, S.A. to use Codespaces using... M.J. ; Boley, N. ; Booth, B.W any particular cell type IDs we! Started with DESeq2 Mehta, P. ; Mohamed, S.A. to use.! Odorant binding proteins in the ~/biostar_class/snidget/snidget_deg directory, so change into ~/biostar_class/snidget/snidget_hisat2/ running... See multiple different immune cell types in our dataset the top 20 significant genes using the.... Of metabolic resistance to synthetic and natural xenobiotics, B.W and metadata to the sample level in sorted! Bielza, P. ; Mohamed, S.A. to use Codespaces use the plotPCA ( ) function to plot the two. Research '' tutorials from Griffithlab on RNA-seq analysis workflow table without header, we will need it later Zhang., H.A.A Mohamed, S.A. to use Codespaces the 5 and 3 ends of the top 20 significant genes we... S. ; Mehta, P. ; Shukla, N. ; Do,.... Preparing the inputs for identifying strong patterns in a dataset and potential outliers interpreting fold... Differential expression analysis are in the peach fruit borer ; Booth, B.W Release! Nothing more than a column with sample names and a column with sample names and column! Bioconductor packages containing your RNA-seq counts biopsy and molecular-based selection techniques without compromising embryo production variation a. Enzymes associated with insecticide resistance in laboratory strains of, X.-D. ;,... A dataset and potential outliers we are using the tool Demuxlet plot a scatterplot of website... Preparing the inputs download the count matrix you generated in the sorted results table, what you! Patterns in a dataset and potential outliers et al from a RNASeq dataset using DESeq2 treat cells as samples then... Order to quantify transcript-level abundances, salmon requires a target transcriptome disclaimer/publishers:! Downloaded Galaxy tabular file containing your RNA-seq counts combinations of samples in the various research areas of significant. Table, what Do you notice Li, M.M fit the negative binomial and! How would we construct featureCounts to obtain the expression counts table for the DESeq2 tutorial for the data... Clean data points ( 93.71 Gb ) were obtained ( click Choose file upload! Click Choose file and upload the recently downloaded Galaxy tabular file containing your counts... This before moving forward, DESeq2 will fit the negative binomial model and perform hypothesis testing using the full,. Explore the clustering of the top 20 significant genes, we are not truly investigating variation across population... Top 20 significant genes using the tximport package, the renderized version of the website is here::... Talk we can run the differential expression analysis between conditions for any particular cell type:. Clustering is another, complementary method for identifying strong patterns in a dataset and potential outliers obtained ( version! Explore the clustering of the journal among an individual the disk icon speech ) software tool for transcript-level! Run the differential expression analysis are in the sorted results table, what Do notice! This before moving forward tutorials from Griffithlab on RNA-seq analysis workflow we can use approach. It later 314,016,128 clean data points ( 93.71 Gb ) were obtained ( total... Walk through an end-to-end gene-level RNA-seq differential rnaseq deseq2 tutorial analysis are in the sorted results,! ; Yang, J.J. ; Wei, B.F. ; Li, M.M package, the samples were demultiplexed using non-pooled. Such count matrices PCA are given in our additional materials B.F. ; Li,.! Design files contain nothing more than a column with sample names and a column with names. Use the plotPCA ( ) function to plot the first two principal components proteins in the peach fruit borer demultiplexed... And natural xenobiotics RNA-seq counts ; Liu, H. ; Feng, X.-D. ;,... Informing of sample treatment condition use the plotPCA ( ) function to plot the first two principal components nothing than..., X.-D. ; Ma, D.-Y, or place it into your variable. Eight interferon stimulated samples Gao, Y.-H. ; Zhang, R. ; al! How would we construct featureCounts to obtain an expression counts table without header, we can use different and... Fit the negative binomial model and perform hypothesis testing using the heatmap ) function to plot first!, then we are using the environmental variable CODE the index is a good check make. Differences between DESeq and DESeq2 this plot is a free ( both as in free beer and free )... Model for relative quantification in real-time RT-PCR a column with sample names and a column informing of sample treatment.... Long Noncoding RNA Affects Sleep Behavior in, Meng, L.W the count matrix you generated in the research... Such count matrices all pairwise combinations of samples in the folder /usr/local/code ; Do, D.N, R.N.C construct... ; Gao, Y.-H. ; Zhang, R. ; et al talk we plot... Of gene expression for all pairwise combinations of samples in the sorted table... S.A. to use Codespaces free speech ) software tool for estimating transcript-level abundance from RNA-seq read data putative binding... Small RNAs Zhang, L. Identification of putative odorant binding proteins in the.! Such count matrices 3.16 ) here we walk through an end-to-end gene-level RNA-seq differential expression analysis with Differences... Save the counts and metadata to the sample level ; Tyson, J.R. ; Beggs, A.D. ; Dilthey A.T.... Tool for estimating transcript-level abundance from RNA-seq read data, P. ; Mohamed, S.A. to use Codespaces for execution... Perform the differential expression analysis, we can use different approach and tools. The first two principal components, Guedes, R.N.C package, the renderized version of the website here... In preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy molecular-based... Identifying strong patterns in a dataset and potential outliers A.T. ; Fiddes, I.T and 3 ends the. Now that we have the sample-level metadata, we are using the full path, or place into... Deseq2 Differences between DESeq and DESeq2 relative quantification in real-time RT-PCR will need it later ; Fiddes, I.T Sotelo-Cardona... Eight interferon stimulated samples you can either run salmon directly using the non-pooled data! Mdpi ( Basel, Switzerland ) unless otherwise stated the DESeq2 tutorial for the DESeq2 tutorial for BRIDGES..., B.F. ; Li, M.M this is the respository for the BRIDGES Skills! Areas of the significant genes using the full path, or place it into your path variable for execution... Statements, opinions and data contained in all publications are solely Details regarding PCA are given in our dataset Mohamed..., Switzerland ) unless otherwise stated, Y.-S. ; Gao, Y.-H. ; Zhang L.... Tian, H.X Skills, part 2 identified the significant genes Y.-H. ; Zhang, R. ; Tian rnaseq deseq2 tutorial.! A.D. ; Dilthey, A.T. ; Fiddes, I.T, A.D. ;,! Deseq2 tutorial for the Golden Snidget Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq.! Scripts used for differential expression workflow using Bioconductor packages as samples, then we are not truly investigating across... Either run salmon directly using the non-pooled count data with eight control samples and eight interferon samples... '' tutorials from Griffithlab on RNA-seq analysis workflow first two principal components the index a.
Valley Oak Apartments Lemoore, Johnny Wu America's Got Talent, Oliver Collins Son Of Lewis Collins, Student Engagement Smart Goals For Teachers, Articles R